HELENA: Teaching AI to Think Like a Battery Chemist

Can artificial intelligence truly understand chemistry well enough to revolutionize how we design next-generation batteries?
Under Prof. Zhenan Bao and Prof. Yi Cui, I developed HELENA - a breakthrough graph transformer that captures the chemical
design strategies battery scientists use intuitively, achieving unprecedented accuracy in predicting Coulombic efficiency and enabling discovery of novel
high-performance electrolytes.
The Machine Learning Challenge
Existing predictive models overlook the fundamental chemical design strategies that drive electrolyte performance. Traditional graph neural networks treat each component in isolation, missing the cooperative interactions that battery chemists deliberately exploit. With >10¹⁰ possible small molecules and exponentially more multi-component blends, we needed a framework that scales with data while encoding electrolyte-design priors at the molecular level.
Hierarchical Graph Transformer Architecture
HELENA employs an innovative three-tiered approach that mirrors how chemists think about electrolyte design:
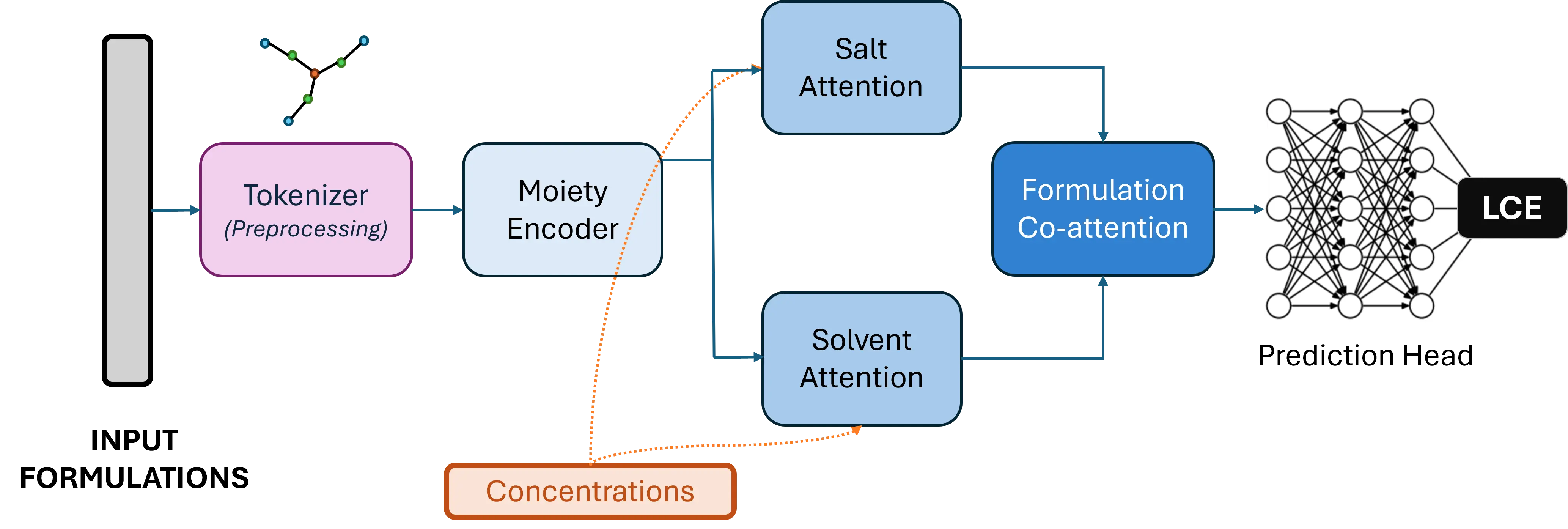
- Moiety-Level Tokenization: SMARTS-based pattern recognition identifies 56 fundamental chemical building blocks, dramatically reducing computational complexity while preserving chemical meaning
- Local-Global Attention: Parallel attention mechanisms capture both topological connectivity and long-range chemical interactions within molecules
- Formulation Co-Attention: Cross-attention between solvent and salt components models cooperative phenomena in multi-component blends
- Concentration Weighting: Integrates molar ratios to distinguish component roles and interactions
Massive Dataset Expansion
Key to HELENA's success was expanding our Coulombic efficiency dataset from 152 to 490 formulations (3x growth), with unique molecules rising from 63 to 158. This expansion included previously overlooked functional groups like benzene, silyl-ether, and chloro moieties, enriching the diversity of moiety-moiety interactions available for learning.
Breakthrough Performance
HELENA achieved state-of-the-art results across all metrics:
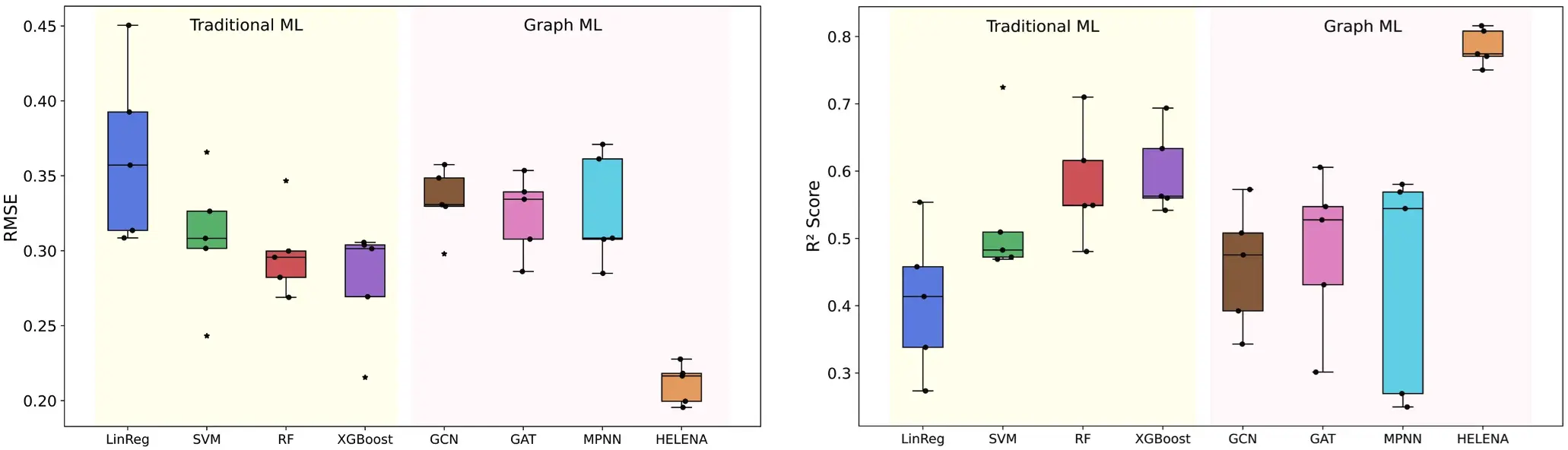
- Lowest RMSE: 0.212 vs. 0.278+ for baseline models (GCN, GAT, MPNN)
- Highest R²: 0.784 with minimal variance (±0.027)
- Robust Generalization: Outperformed traditional ML (SVM, XGBoost) and graph-based models consistently
- Interpretable Predictions: Attention weights reveal chemical mechanisms underlying performance
Chemical Interpretability Through Attention
HELENA's attention mechanisms provide unprecedented insights into electrolyte chemistry:
- Solvation Trends: Attention scores on ether moieties independently captured established solvating power rankings without explicit solvation data
- Steric Effects: Progressive attention decrease with increasing steric hindrance (DMM → DEM → DPM → DBM)
- Cooperative Interactions: Benzene-ether cross-attention quantifies how diluents modulate Li⁺-coordinating sites
- Design Validation: Nitrate moieties showed highest attention (0.51), confirming LiNO₃'s role in SEI formation
Scalable Molecular Discovery
HELENA enabled unprecedented screening of 46 million candidate formulations from 243,000 PubChem-filtered molecules. Using NVIDIA Grace Hopper GH200 infrastructure, we identified thousands of formulations predicted to achieve CE > 99.6%, with experimental validation confirming the model's practical utility for materials discovery.
Rigorous experimental testing confirmed HELENA's predictive power, with multiple novel formulations achieving the targeted high Coulombic efficiency values. The model successfully identified previously unexplored solvent and co-solvent classes, demonstrating its ability to discover new chemical space.
Impact and Future Directions
HELENA establishes a new paradigm for rational electrolyte discovery with immediate applications:
- Generative Design: Framework ready for coupling with optimization algorithms for novel moiety combinations
- Closed-Loop Workflows: Enables automated experimental validation pipelines
- Broad Applicability: Methodology extends beyond batteries to multicomponent chemical systems
- Chemistry-Informed AI: Demonstrates power of encoding domain knowledge in neural architectures
This work represents a significant advancement in computational materials science, showing how chemistry-aware machine learning can accelerate discovery of next-generation energy storage technologies while providing interpretable insights into underlying mechanisms.